Der „Periodic Safety Update Report (PSUR)“ – Ein neuer Spieler auf dem Feld
Ein wesentliches Ziel der Neuordnung der Zugangsvoraussetzungen für Medizinprodukte im europäischen Wirtschaftsraum durch die EU-Verordnungen 2017/745/EU (MDR) ist die Gewährleistung der Produktsicherheit über den gesamten Lebenszyklus.
Medizinproduktesicherheit als Grundpfeiler der MDR
Schon in den Erwägungsgründen zur MDR stellen das europäische Parlament und der Rat der europäischen Union dar, dass Hersteller verpflichtet sind, mit und über das Qualitätsmanagementsystem ein Instrumentarium einzurichten, welches es erlaubt, das Verhalten eines Medizinprodukts in der regulären Anwendung zu überwachen. Dieses System zur Post-Market Surveillance (PMS; System zur Überwachung nach dem Inverkehrbringen) hat zum Ziel, über notwendige Maßnahmen zu entscheiden, um die Sicherheit der Patienten und Anwender zu gewährleisten. Die Datengrundlage dazu wird über die systematische und aktive Erfassung von Anwendungserfahrungen aus verschiedensten Quellen geschaffen.
Der „PSUR“ als zentrales Element der PMS
Um einen effizienten Prozess zu schaffen, müssen die einzelnen Elemente der PMS in ihren Aufgaben klar abgegrenzt sein. Zudem müssen Informationskanäle geschaffen werden, um das erfasste Wissen zu bündeln und an den richtigen Stellen zu verarbeiten. Der PSUR steht dabei an zentraler Position. In diesem werden die zentralen, aktuellen PMS-Erkenntnisse einer Berichtsperiode (mindestens zweijährlich bei Klasse IIa; ab Klasse IIb/Implantate jährlich) zusammengefasst und analysiert.
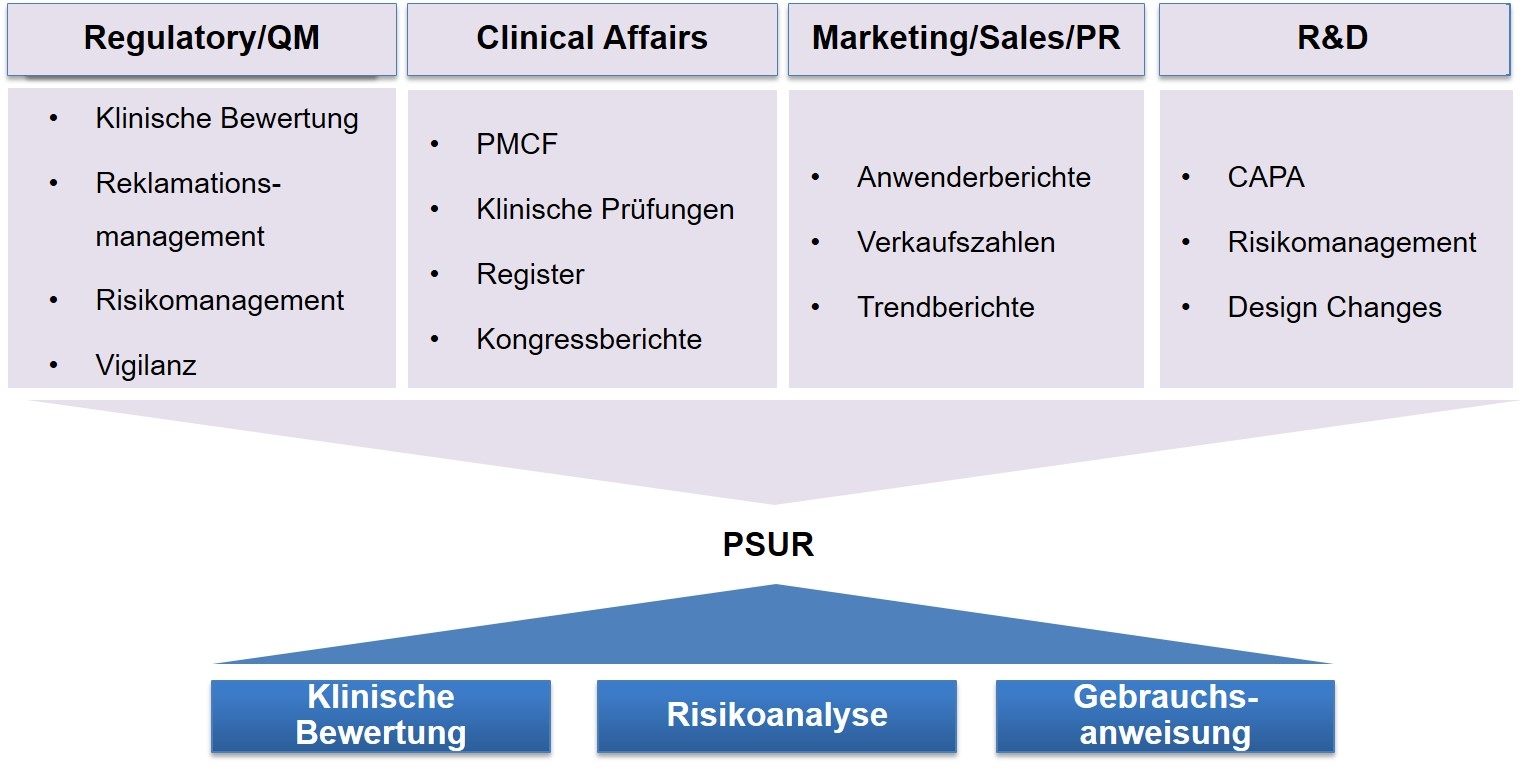 Die Etablierung eines PSUR erfordert die Integration verschiedener Informationslinien in einen Prozess, daher den Aufbau eines „Wissensmanagements“.
© novineon Healthcare Technology Partners GmbH/novineon CRO & Consulting Ltd
Die Etablierung eines PSUR erfordert die Integration verschiedener Informationslinien in einen Prozess, daher den Aufbau eines „Wissensmanagements“.
© novineon Healthcare Technology Partners GmbH/novineon CRO & Consulting Ltd
Der PSUR soll klar strukturiert aufgebaut sein, um einen Abgleich der Erkenntnisse mit der vorhandenen Wissensbasis (dargelegt in der Risikoanalyse und der klinischen Bewertung) zu ermöglichen. Daher soll der PSUR auch klar von diesen Dokumenten abgegrenzt sein.
Grundlagen einer sinnvollen PMS-Strategie
Ein wichtiger Bestandteil der PMS ist das Zusammentragen sowie die Auswertung von Informationen und Reklamationen durch Kunden/Anwender, der Rückmeldungen des Vertriebs (= z. B. durch die Medizinprodukteberater) und/oder des Wartungsdienstes sowie interner Prüfergebnisse. Neben der Beobachtung des eigenen Produkts gehört auch die Beobachtung von Mitbewerberprodukten zu einer MDR-konformen PMS-Strategie. Das gilt insbesondere in solchen Fällen, in denen die Klinische Bewertung des eigenen Produkts auf dem Literaturweg über die Auswertung klinischer Daten von Mitbewerberprodukten basiert. Ein systematisches PMS-Verfahren beinhaltet weiterhin die Auswertung von Behördenmeldungen (BfArM, MAUDE-Datenbank) sowie die Auswertung der wissenschaftlichen Literatur über das eigene Produkt und Mitbewerberprodukte.
Die europäische Medizinprodukterichtlinie 93/42/EWG verpflichtete Hersteller zwar, Überwachungsprogramme innerhalb ihres Qualitätsmanagementsystems zu implementieren, die routinemäßig die klinische Wirksamkeit und Sicherheit des betreffenden Produkts am Markt und in der Anwendung überprüft. Über die genaue Ausgestaltung solch eines PMS-Verfahrens gab die europäische Medizinprodukterichtlinie allerdings keine spezifizierten Vorgaben. Es lag und liegt in der Herstellerverantwortung ein dem Produkt angepasstes PMS-Konzept zu entwickeln.
Diese Vorgaben werden nun durch die MDR in einem eigenen Kapitel VII plus Anhang III konkretisiert und in ihrer Wertigkeit deutlich gesteigert.
Der Artikel 83 fordert ein PMS-System, das als Teil des QM-Systems
- aktiv
- systematisch
- kontinuierlich
Daten über die Qualität, die Leistung und die Sicherheit eines Produkts sammelt und bewertet. Hersteller mit einem geeigneten und durchdachten Qualitätsmanagement haben auch unter den neuen Rahmenbedingungen nichts zu befürchten. Die zusätzlichen Anforderungen des Artikel 83 nach solch einem PMS-System verlangen aber nach einer Anpassung, die sich in gute QM-Systeme implementieren lassen sollten. Hersteller, welche der schon unter der 93/42/EWG bestehenden Verpflichtung zur PMS konsequent nachkommen, werden sich auch mit den neuen Regelungen arrangieren können.
So fordert die MDR die Etablierung neuer Instrumente, die in die bestehenden Prozesse und Beziehungen zwischen PMS/PMCF, klinische Bewertung und Risikomanagement einzupassen und abzugrenzen sind. Kernelemente bestehen, neben der Errichtung eines PMS-Systems, auch in erweiterten Dokumentations- und Berichtspflichten wie dem PMCF-Plan zur klinischen Nachbeobachtung (Post-Market Clinical Follow-up; zur klinischen Nachbeobachtung) und dem PMCF-Bericht nach Artikel 61 sowie dem PMS-Plan (Artikel 84; Plan zur Überwachung nach dem Inverkehrbringen) mit dem PMS-Report (Bericht über die Überwachung nach dem Inverkehrbringen; Artikel 85 für Klasse-I-Produkte) bzw. PSUR (Regelmäßig aktualisierter Bericht über die Sicherheit, Artikel 86 für Klasse IIa und höher) und nicht zuletzt mit dem SSCP (Kurzbericht über Sicherheit und klinische Leistung; Artikel 32 für Klasse-III-Produkte/Implantate). In der Gesamtheit stellen diese Prozesse/Berichte die Datenbasis zur Sicherstellung der Sicherheit und Leistung der in Verkehr gebrachten Medizinprodukte.
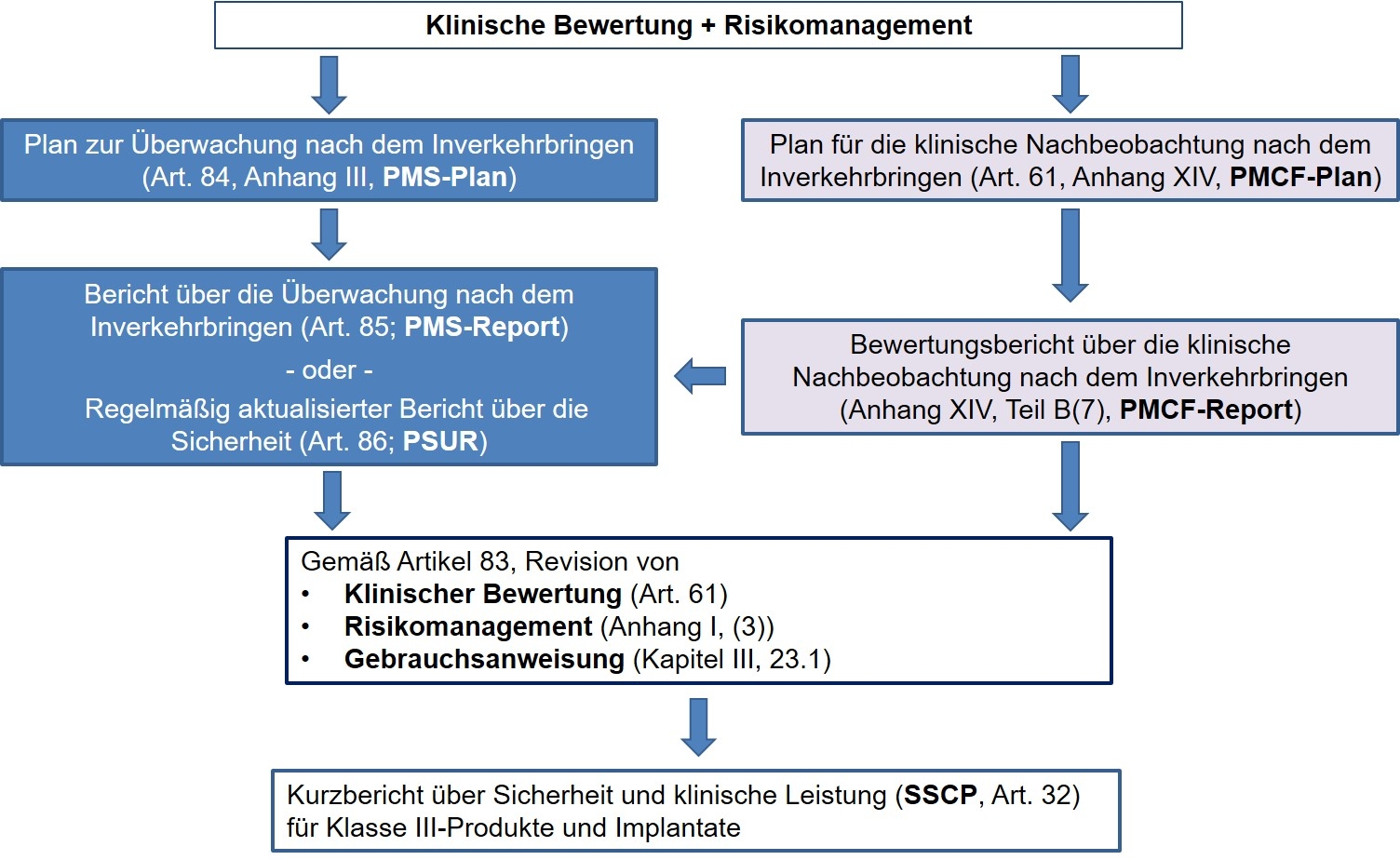 Flussschema des gesamten PMS-Prozesses, abgeleitet aus den Forderungen der MDR
© novineon Healthcare Technology Partners GmbH / novineon CRO & Consulting Ltd
Flussschema des gesamten PMS-Prozesses, abgeleitet aus den Forderungen der MDR
© novineon Healthcare Technology Partners GmbH / novineon CRO & Consulting Ltd
Initial, daher idealerweise begleitend zum Entwicklungsprozess und verpflichtend im Rahmen des Konformitätsbewertungsverfahrens, wird das Nutzen-Risiko-Verhältnis eines Medizinprodukts im Rahmen einer Risikoanalyse und klinischen Bewertung erfasst. Diese Prozesse erfassen den gegebenen Stand der Erkenntnis sowie die prospektiv erstellten Annahmen über Risiken in Verbindung mit der Anwendung des Produkts. Dennoch beruht letztlich jedes Konformitätsbewertungsverfahren mehr oder weniger auf einer Momentaufnahme zum Zeitpunkt der Erstellung. Trotz bester Ableitung von allgemeinem und speziellem Wissen über das Produkt, die Produktklasse und das medizinische Anwendungsumfeld muss sich ein Medizinprodukt „in realitas bewähren“. Die im Rahmen der klinischen Bewertung und klinischen Prüfung erkannten Restrisiken sowie das Nutzen-Risiko-Verhältnis müssen daher ständig mit Erkenntnissen aus der Anwendung neu bewertet werden.
Der Aufbau der dazu notwendigen Datengrundlage ist, neben der nachfolgenden Ableitung von Maßnahmen zur Gewährleistung der Sicherheit die Aufgabe des gesamten PMS-Prozesses.
Proaktives Management von Wissen
Die klinische Bewertung/Risikoanalyse stellt den kumulierten Erkenntnisstand / die Wissensbasis der vorhergegangenen Berichtsperioden dar. Der PSUR (gefüttert aus den aktuellen PMS/PMCF-Erfahrungen) dient dem periodischen Abgleich der tradierten Sicherheitserkenntnisse mit neuen Erkenntnissen sowie den eingeleiteten Maßnahmen (sofern vorhanden).
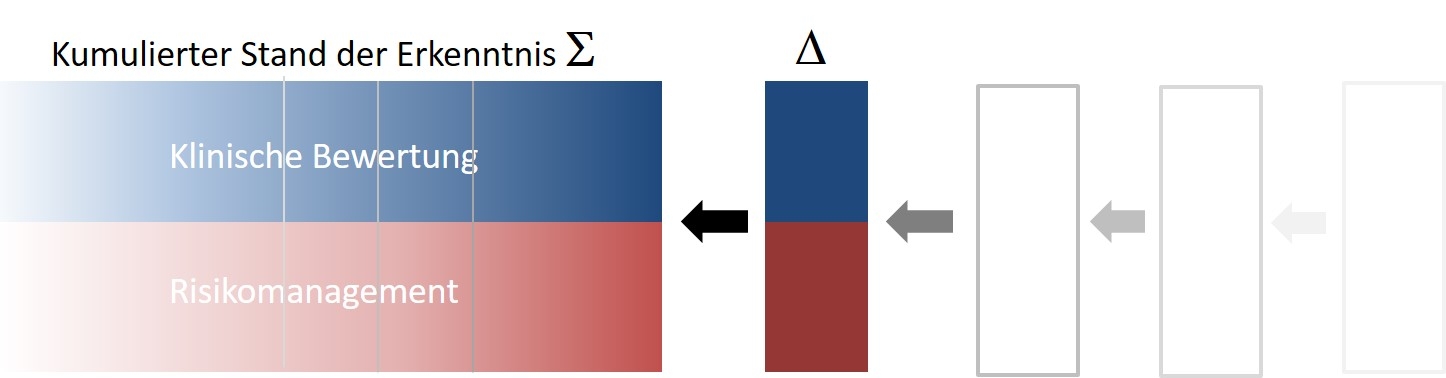 Ausgehend von diesem Modell liegt der Fokus des PSUR auf der aktuellen Berichtsperiode und kann, je nach neuer Erkenntnislage, schlank gehalten werden.
© novineon Healthcare Technology Partners GmbH / novineon CRO & Consulting Ltd
Ausgehend von diesem Modell liegt der Fokus des PSUR auf der aktuellen Berichtsperiode und kann, je nach neuer Erkenntnislage, schlank gehalten werden.
© novineon Healthcare Technology Partners GmbH / novineon CRO & Consulting Ltd
Adressaten des PSUR sind in unterschiedlichen Stufen der Berichtspflicht die Benannte Stelle sowie die zuständigen Behörden. Um eine einheitliche Bewertung und eine klare Übersichtlichkeit zu erreichen, sollen standardisierte Berichtsvorlagen bzw. Richtlinien durch den Gesetzgeber erstellt werden.
Die Anstrengung liegt für die Hersteller nicht so sehr in der eigentlichen Erstellung des PSUR, der trotz seines neuartigen Auftretens in der MDR keine neuen Inhalte fordert. Die Schlussfolgerungen aus der Nutzen-Risiko-Bewertung, die Erkenntnisse der PMCF sowie quantitative Angaben zu Absatz- und Anwendungsmengen müssen schon aus den unter der Richtlinie 93/42/EWG geforderten Mechanismen zur Verfügung stehen.
Dennoch müssen Hersteller die Prozesse und Datenströme schaffen, um die konkretisierten PMS-Anforderungen zu erfüllen und zu validen Schlussfolgerungen zu gelangen. Ein durchdacht erstellter und konsequent umgesetzter PMS-Plan bestätigt die Sicherheit und Leistungsfähigkeit von Medizinprodukten bzw. identifiziert früh mögliche Sicherheitsproblematiken und definiert geeignete Maßnahmen.
Anmerkung der Redaktion:
Die Veröffentlichung dieses Beitrages erfolgt mit freundlicher Genehmigung der novineon Healthcare Technology Partners GmbH/novineon CRO & Consulting Ltd.