Experteninterview
Die neue EU-Verordnung über In-vitro-Diagnostika (IVDR) – Herausforderungen und Chancen für Hersteller
Die neue EU-Verordnung über In-vitro-Diagnostika (IVDR) ist am 25. Mai 2017 mit einem Übergangszeitraum von fünf Jahren in Kraft getreten und findet ab dem 26. Mai 2022 in vollem Umfang Anwendung. Die vielen Vorteile, die die neue Verordnung mit sich bringen soll, wie die Erschaffung eines transparenten international anerkannten Rechtsrahmens und die Verbesserung der klinischen Sicherheit, gehen gleichzeitig mit großen Herausforderungen für die Hersteller von In-vitro-Diagnostika (IVD) einher. Vor allem für Kleinst- und Kleinunternehmen bedeutet die Umsetzung der neuen Verordnung einen großen zeitlichen Mehraufwand und hohe Kostenintensität. Die wichtigsten Erkenntnisse zu den kommenden Herausforderungen wie auch mögliche Chancen erklärt der Experte Dr. Sebastian Grömminger vom Johner Institut im Interview mit Amina Daca von der BIOPRO Baden-Württemberg GmbH.
 Dr. Sebastian Grömminger unterstützt im Johner Institut die Kunden bei der Zulassung von In-vitro-Diagnostika.
© Johner Institut
Dr. Sebastian Grömminger unterstützt im Johner Institut die Kunden bei der Zulassung von In-vitro-Diagnostika.
© Johner Institut
Eine der wichtigsten Veränderungen, welche die IVDR mit sich bringt, betrifft die Risikoklassifizierung der Produkte. Was ändert sich mit dem neuen regelbasierten Klassifizierungssystem grundlegend für den Hersteller?
Eine der bedeutendsten Änderungen ist, dass Hersteller mit Produkten ab Klasse B und höher künftig von einer Benannten Stelle überwacht werden. Somit steigt der Anteil an überwachten Produkten und Herstellern von 10-15 % unter IVD-Richtlinie nun auf 85-90 % unter der IVD-Verordnung (IVDR). Zudem wird der Dokumentationsumfang stark steigen und die Dauer bis zur Markteinführung eines Produktes wird sich verlängern.
Wie legt man als Hersteller die Zweckbestimmung eines Produktes fest? Und wie geht man am besten vor, wenn mehrere Zweckbestimmungen vorliegen?
Die Zweckbestimmung festzulegen ist nicht einfach, vor allem, wenn mehrere Disziplinen involviert sind, wie Assay, Gerät und/oder Software, die im Zusammenspiel erst ein Ergebnis liefern. Dann ist es eine strategische Entscheidung, die Produkte zu trennen oder sie zusammen als ein Produkt zu beschreiben.
Hersteller sollten präzise in der Zweckbestimmung formulieren, was das Produkt können soll und in welchem Kontext. So sollte bereits an der Zweckbestimmung erkennbar sein, ob das Produkt für den professionellen Einsatz im Labor, als patientennahe Lösung (Point-of-Care-Testing) oder gar für die Eigenanwendung gedacht ist. Der Probentyp sollte spezifiziert sein, wie zum Beispiel Blut, Urin oder Speichel. Falls bereits vorbehandeltes Probenmaterial wie zum Beispiel gelöste Nukleinsäuren in wässriger Pufferlösung verwendet werden sollen, so sollte die Zweckbestimmung dies darlegen.
Ähnliche Zweckbestimmungen von mehreren Produkten können in sogenannte „generische Produktgruppen“ zusammengefasst werden. Wenn mehrere Zweckbestimmungen bei einem einzelnen Produkt vorliegen, fällt das gesamte Produkt in die höchste Klasse, die beim Durchlaufen der Klassifizierungsregeln ermittelt wird. Es ist somit ratsam zu prüfen, ob eine Auftrennung des Produktes basierend auf den verschiedenen Zweckbestimmungen hilfreich ist, um Teile des bisherigen Produktes getrennt zu klassifizieren.
Gibt es Guidelines, die den Herstellern die Einstufung der einzelnen Produkte in die Risikoklassen erleichtern können?
Wir warten noch immer auf die „Durchführungsrechtsakte“ oder MEDDEV Guidances, die Hilfestellung bei der Klassifizierung bieten könnten. Die Klassifizierungsregeln der IVDR basieren auf einem Guidance-Dokument des International Medical Device Regulators Forum, IMDRF (ehemals GHTF, Global Harmonization Task Force) mit dem Titel “Principles of In Vitro Diagnostic (IVD) Medical Devices Classification“ vom 19. Februar 2008 (Kennnummer GHTF/SG1/N045:2008). In diesem Dokument finden sich für jede Regel auch Begründungen, die es erlauben, die Intention hinter den Klassifizierungsregeln zu verstehen.
Das Konformitätsbewertungsverfahren eines Produktes für die CE-Kennzeichnung hängt von der Risikoklasse und den besonderen Merkmalen des Produkts ab. Was sollte der Hersteller bei der Wahl des Verfahrens zur Bewertung der Konformität beachten?
Hersteller sollten ein Konformitätsbewertungsverfahren wählen, das zu den geplanten Update- bzw. Änderungszyklen passt. Wenn ein Produkt seit vielen Jahren unverändert ist und auch unter der IVDR voraussichtlich nur sehr selten bis gar nicht über den Produktlebenszyklus hinweg geändert wird, mag die Wahl der Konformitätsbewertung über Anhang X und XI, die sogenannte „Baumusterprüfung“, gepaart mit der Qualitätssicherung der Produktion sinnvoll sein. Sobald jedoch in absehbarer Zeit auch unwesentliche Änderungen bevorstehen – und das betrifft vor allem Produkte, die Software enthalten oder aus Software bestehen, rate ich zum Konformitätsbewertungsverfahren nach Anhang IX (Qualitätsmanagementsystem und Prüfung der technischen Dokumentation).
Was ändert sich mit der IVDR zukünftig in der Zusammenarbeit mit den Benannten Stellen?
Die Benannten Stellen werden sehr viel mehr Produkte bewerten müssen, wodurch vorhersehbar mit Engpässen bei Benannten Stellen zu rechnen ist. Die Benannten Stellen bauen zwar Personalressourcen auf, die aber erst geschult werden müssen. Dieses theoretische Wissen wird dann direkt auf die Praxis angewandt. Das kann im Einzelfall bedeuten, dass eine wortgenaue Auslegung der IVDR die Basis einer Dokumentenprüfung oder eines Vor-Ort-Audits die Folge sind. Je unpräziser die Dokumentation vorliegt, desto eher kann es dann zu Abweichungen im Konformitätsbewertungsverfahren kommen.
Mit zunehmender Risikoklasse eines Produkts wird der Umfang des klinischen Nachweises, der erforderlich ist, um die Konformität des Produktes zu belegen, immer strenger. Worauf beruht der klinische Nachweis eines In-vitro-Diagnostikums und was ändert sich mit der IVDR?
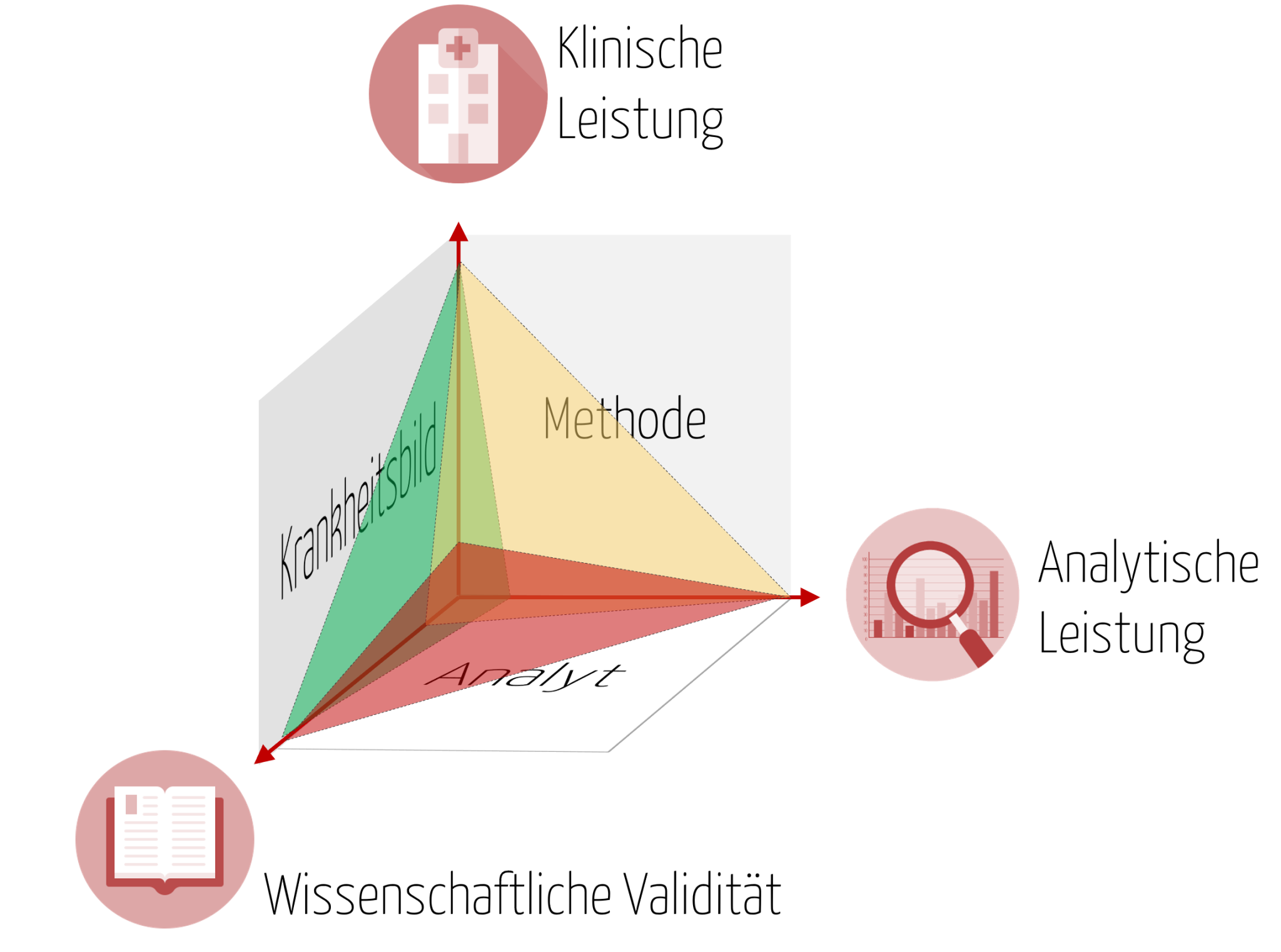 Magisches Dreieck zur Leistungsbewertung eines In-vitro-Diagnostikums
© Johner Institut / Dr. Sebastian Grömminger
Magisches Dreieck zur Leistungsbewertung eines In-vitro-Diagnostikums
© Johner Institut / Dr. Sebastian Grömminger
Der klinische Nachweis ist erforderlich, um den klinischen Nutzen für ein diagnostisches In-vitro-Medizinprodukt zu belegen. Dieser klinische Nutzen stellt die positiven Auswirkungen eines Produktes im Zusammenhang mit seiner Funktion dar, also zum Beispiel Überwachung, Diagnose, das Patientenmanagement oder gar die öffentliche Gesundheit. Je nach Risikoklasse ist es besonders wichtig den klinischen Nachweis zu erbringen, damit der Nutzen des Produktes fortlaufend belegt werden kann. Denn sobald die Leistung des Produktes auf analytischer und klinischer Ebene nicht oder nicht mehr dem allgemein anerkannten Stand der Technik entspricht bzw. durch neue wissenschaftliche Erkenntnisse überholt wurde, stellt das Produkt ggf. ein nicht mehr vertretbares Risiko für Patienten dar.
Mit der IVDR wird nun ein kontinuierlicher Prozess zur Leistungsbewertung gefordert, der immer wieder eine Neubewertung des, wie ich es nenne „magischen Dreiecks“ fordert, bestehend aus 1.) wissenschaftlicher Validität des Analyten, 2.) analytischer Leistung und 3.) klinischer Leistung, auch wenn das Produkt bereits seit langem in Verkehr gebracht und auf dem Markt verfügbar ist.
Welche Auswirkungen hat die neue IVDR auf die Zusammenarbeit zwischen Original Equipment Manufacturer (OEM) und Private Label Manufacturer (PLM)?
Die PLM-OEM-Konstellationen wird es in der bisherigen Form nicht mehr geben, da die Wirtschaftsakteure insgesamt deutlich stärker in die Verantwortung genommen werden und infolge dessen stärker miteinander zusammenarbeiten müssen. Im Falle von PLM muss ein Vollzugriff auf die technische Dokumentation des OEM gewährleistet sein. Konflikte zwischen OEM und PLM, die sich um das geistige Eigentum drehen, sind somit in den meisten Fällen vorprogrammiert. Oftmals ist es ratsam, dass der bisherige PLM zum Distributor wird. Dies sollte jedoch im Einzelfall genau beleuchtet werden, ob dies für die jeweilige Geschäftsgrundlage zielführend ist.
Welche Möglichkeiten zur Förderung gibt es für KMUs, um die Kosten für die klinische Validierung zu bewältigen? Gibt es Unterstützung für die regulatorischen Anforderungen?
Ja, das BMBF bietet zum Beispiel Förderprogramme genau für diese Zwecke. Für die Unterstützung hinsichtlich regulatorischer Anforderungen sind mir bisher keine Förderungen bekannt. Es gibt jedoch einige Veranstaltungen, wie zum Beispiel von BIOPRO. Hier bietet sich die Möglichkeit mit Fachexperten Kontakt aufzunehmen und das ein oder andere Thema kostenlos anzusprechen. Wir vom Johner Institut bieten sehr viele kostenlose Informationen auf unserem Blog und im Newsletter sowie ein kostenloses Microconsulting. Hierbei haben wir in den letzten 20 Monaten bereits über 2.900 Fragen meist binnen eines Arbeitstages beantwortet.
Die Bedürfnisse der Hersteller bei der Anpassung ihrer Produkte an die IVDR sind individuell. Die einen müssen sich mit PLM-OEM-Fragestellungen auseinandersetzen, die anderen müssen die Wiederaufbereitung der Produkte angehen. Zu welchem Vorgehen würden Sie Herstellern bei spezifischen Fragestellungen raten?
Das Wichtigste ist in meinen Augen, einen konkreten Aktionsplan für das eigene Unternehmen zu erstellen. Die höchste Priorität hat aktuell die Fortbildung des Personals bzw. bestimmter Schlüsselrollen, da dies die ressourcenschonenste Maßnahme darstellt. Die Überprüfung der Zweckbestimmung würde ich als die Basisentscheidung für den Aktionsplan betrachten, bzw. ob ein PLM weiterhin als legaler Hersteller in Erscheinung treten möchte und darf. Anschließend sollte die Klassifizierung der Produkte vorgenommen werden. Sobald dann Klasse B oder höher das Ergebnis ist, wird es höchste Zeit, sich eine Benannte Stelle zu suchen. Denn ansonsten kann es dazu kommen, dass man, abgesehen von Übergangsbestimmungen in Artikel 110, bis 26. Mai 2022 keine Marktzugangsberechtigung mehr hat. Erst wenn diese Punkte geklärt sind, sollten sich Hersteller den einzelnen Fragestellungen im Detail widmen, es sei denn, es sind ausreichend personelle und/oder finanzielle Ressourcen vorhanden, die eine Parallelisierung der Aktivitäten ermöglichen.